|
Jenti
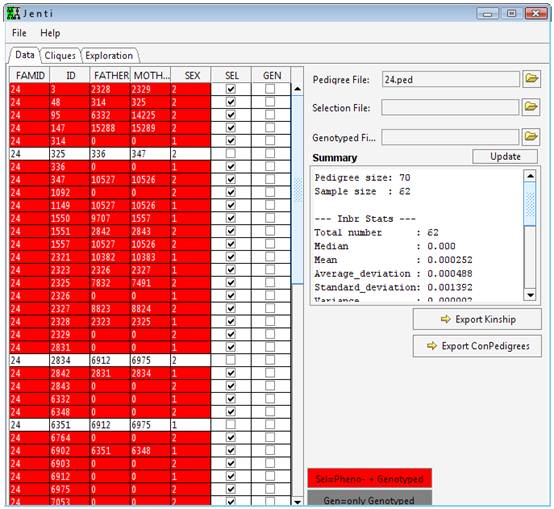
requires a file describing the genealogical data and a selection flag
indicating which individuals will be considered in the clustering process
and/or in the optional sub-pedigree reconstruction. A separate list of
individuals could be also provided in separate files to overriding the
selection flags.
The
pedigree file describes the relationships between individuals using a
standard format. Each row of the file describes a single individual and
has the following standard format:
GenealogyID PersonID FatherID MotherID Sex
SelectionFlag
The
GenealogyID is a numeric code,
unique within the genealogy.
The
PersonID is a numeric code
that uniquely identifies an individual.
FatherID
and MotherID are the numeric
codes that specify the individuals parents. If an individual is a
founder each parent identifier field contains a 0, otherwise the two
parents should be present in the same family.
The
Sex field contains 1 for a male or 2 for a female.
The
SelectionFlag field contains
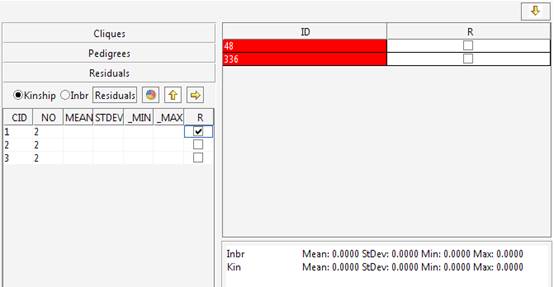
2 if the individual must be included in the clustering process analysis
or 0 if the individual must not be included in the cluster. If the aim
of the study is generating sub-pedigrees suitable for family-based
studies we would like to include in the sub-pedigrees all the
first-degree related individuals having genotype data. This information
is provided setting the SelectionFlag
to 1.
All
fields must be separated by a single TAB or by comma.
Example:
parents and two sons. The sons will be included in the clustering
process, the mother has genotypic data.
1 1 0 0 1 0
1 2 0 0 2 1
1 3 1 2 1 2
1 4 1 2 1 2
etc
The
default extension for a genealogy file is ped.
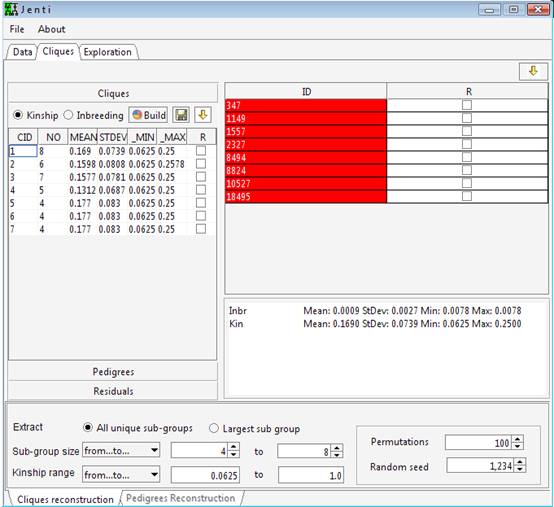
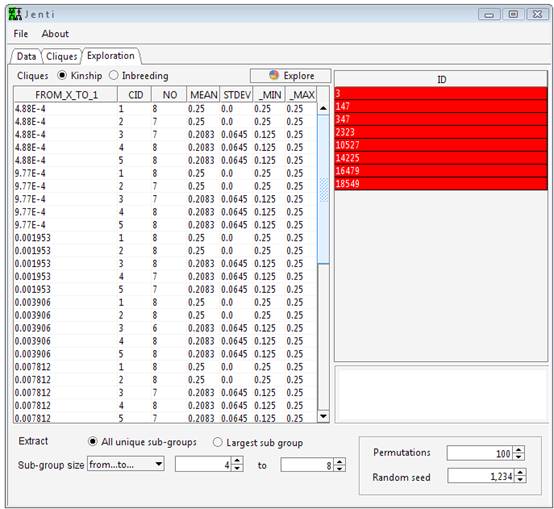
Optionally,
the lists of individuals to be included in the clustering process or in
the sub-pedigree reconstruction step could be provided in separated files
listing one PersonID on each
line. These data will overwrite the information given in the genealogical
file and allows using a single genealogical file for different studies.
Attention PersonID,FatherID and MotherID have to be numeric and not larger than 32000. Thanks to Clemens Egger we can provide an en/decoding tool.
|